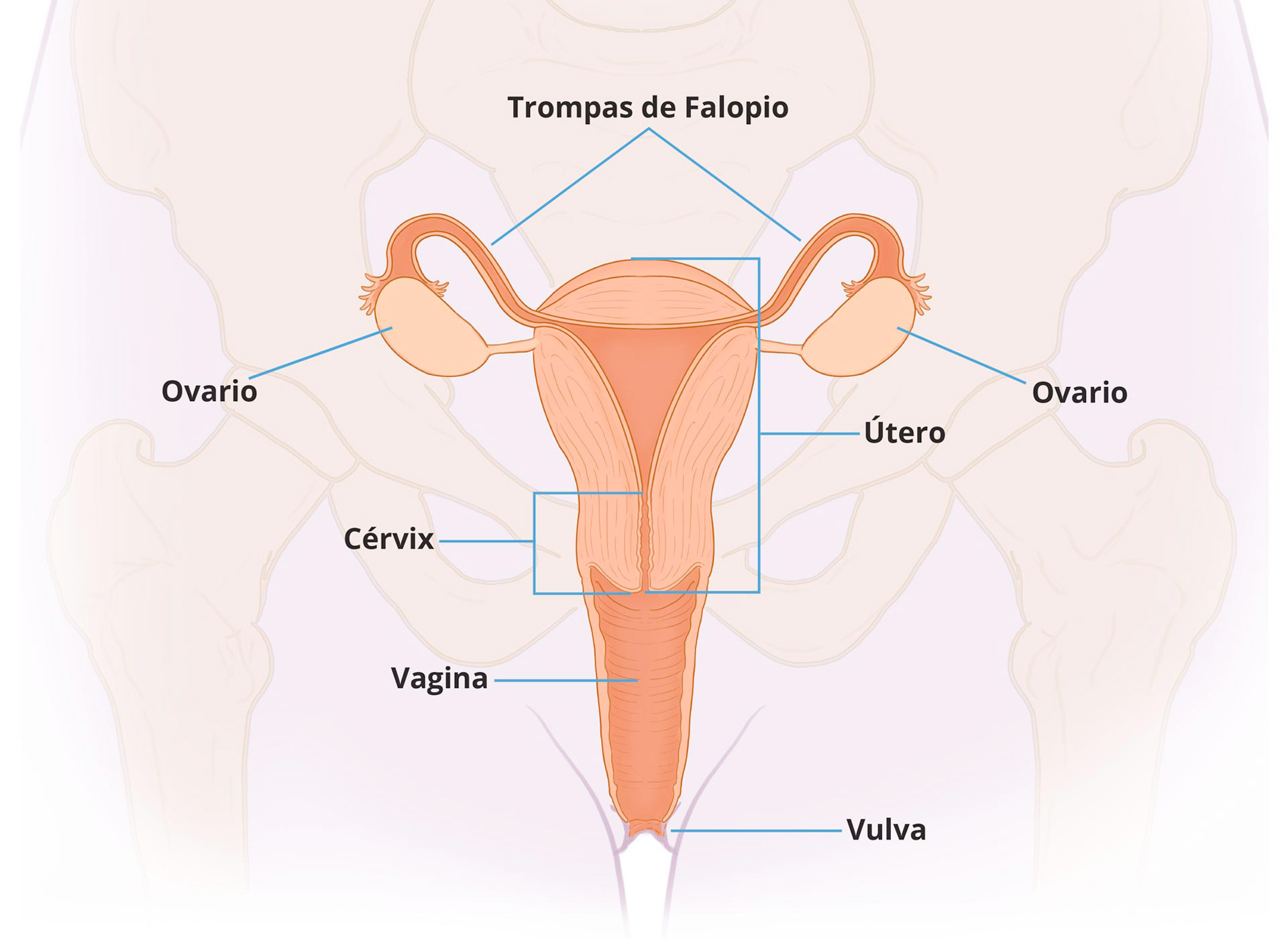
Los estrógenos y la progesterona son las hormonas sexuales femeninas. Se producen en el ovario y controlan básicamente los ciclos menstruales produciendo cambios en el endometrio, que lo prepararán para recibir un posible embarazo.
ESTRÓGENOS
Química y síntesis
Química. Muchos compuestos esteroideos y no esteroideos, poseen actividad estrogénica. Los estrógenos interactúan con dos receptores de la superfamilia de receptores nucleares, denominados ERα y ERβ. El estrógeno natural más potente en humanos para las acciones mediadas por ERα como ERβ, es 17β-estradiol, seguido por estrona y estriol. Cada uno contiene un anillo fenólico A, con un grupo hidroxilo en el carbono 3, y un β-OH o cetona, en la posición 17 del anillo D. El anillo fenólico A es la principal característica estructural responsable de la unión selectiva, de alta afinidad, a ambos receptores. La mayoría de las sustituciones de alquilo en el anillo A dificultan la unión, pero pueden ser toleradas las sustituciones en el anillo C o D. Las sustituciones de etinilo en la posición C17 aumentan, en gran medida, la potencia para producir los efectos deseados por vía oral, al no experimentar metabolismo hepático de primer paso. Los modelos para los sitios de unión a ligando de ambos ER se han determinado a partir de relaciones estructura-actividad y análisis estructural. Los ligandos selectivos para ERα y ERβ están disponibles para estudios experimentales, pero aún no se usan terapéuticamente.
Biosíntesis. Los estrógenos esteroideos se originan de la androstenediona o la testosterona por la aromatización del anillo A. La reacción es catalizada por la aromatasa (CYP19), que utiliza NADPH y oxígeno molecular como cosustratos. También es esencial una flavoproteína omnipresente, NADPH-citocromo P450 reductasa. Ambas proteínas están localizadas en el retículo endoplasmático de células de la granulosa ovárica; en células testiculares de Sertoli y Leydig, en estroma adiposo, sincitiotrofoblastos placentarios, en blastocistos de preimplantación, huesos, diversas regiones cerebrales y en muchos otros tejidos. Los ovarios son la fuente principal de estrógenos circulantes en mujeres premenopáusicas, siendo el estradiol el principal producto secretor. Tradicionalmente, se piensa que la producción de estradiol ovárico requiere de dos tipos de células: células de teca y células de granulosa. La gonadotropina LH actúa, a través de receptores que se acoplan a la ruta de AMP cíclico-adenilil ciclasa, para aumentar el transporte de colesterol (precursor de todos los esteroides) en las mitocondrias de las células, donde se producen los precursores andrógenos. La FSH estimula entonces, la producción y actividad de CYP19 en las células de la granulosa, que convierte los precursores andrógenos en estrógenos. Notablemente, las células de teca del ovario contienen una forma de 17β-hidroxiesteroide deshidrogenasa (tipo I) que favorece la producción de testosterona y estradiol, a partir de androstenediona y estrona, respectivamente. Sin embargo, en el hígado, otra forma de esta enzima (tipo II) favorece la oxidación del estradiol circulante a estrona, y ambos esteroides se convierten luego en estriol. Los tres estrógenos se excretan en la orina, junto con sus conjugados de glucurónido y sulfato.
En las mujeres posmenopáusicas, la principal fuente de estrógeno circulante es el estroma del tejido adiposo, donde la estrona se sintetiza a partir de deshidroepiandrosterona, secretada por las glándulas suprarrenales. En los hombres, los estrógenos son producidos por los testículos, pero la producción extragonadal por aromatización de los esteroides C19 circulantes (p. ej., androstenediona y deshidroepiandrosterona) representa la mayoría de los estrógenos circulantes.
Acciones fisiológicas
Acciones de desarrollo. Los estrógenos son responsables en gran medida de los cambios puberales en las niñas y la aparición de las características sexuales secundarias. Los estrógenos causan el crecimiento y el desarrollo de la vagina, el útero y las trompas de Falopio, y contribuyen al aumento de volumen de los senos. También contribuyen a moldear los contornos del cuerpo, dar forma al esqueleto y provocar el crecimiento puberal de los huesos largos y el cierre epifisiario. El crecimiento del vello axilar y púbico; la pigmentación de la región genital y la pigmentación regional de los pezones y las areolas, que ocurren después del primer trimestre del embarazo, también son acciones estrogénicas. Los andrógenos también pueden desempeñar un papel secundario en el desarrollo sexual femenino.
Control neuroendocrino del ciclo menstrual

El control del ciclo menstrual se lleva a cabo mediante una cascada de señales neuroendocrinas que involucran al hipotálamo, la hipófisis y los ovarios. Un oscilador neuronal, o reloj, se dispara en el hipotálamo a intervalos que coinciden con descargas de GnRH liberada en la vasculatura portal hipotalámica-hipofisiaria. GnRH interactúa con su receptor análogo, en gonadotropos hipofisiarios causando la liberación de LH y FSH. La frecuencia de los pulsos de GnRH, que varía en las diferentes fases del ciclo menstrual, controla la síntesis relativa de las subunidades β, en particular de FSH y LH. Las gonadotropinas (LH y FSH) regulan el crecimiento y la maduración del folículo de Graaf en el ovario, y la producción ovárica de estrógenos y progesterona, que ejercen una regulación de retroalimentación sobre la hipófisis y el hipotálamo. Debido a que la liberación de GnRH es intermitente, la secreción de LH y FSH es pulsátil. La frecuencia del pulso está determinada por el reloj neural, denominado generador hipotalámico de pulsos GnRH, pero la cantidad de gonadotropina liberada en cada pulso (es decir, la amplitud del pulso) está en gran parte controlada en la glándula hipofisiaria por las acciones de los estrógenos y la progesterona. La naturaleza pulsátil y de intermitencia de la liberación hormonal es esencial para el mantenimiento de los ciclos menstruales ovulatorios normales, porque la infusión constante de Gn-RH produce el cese de la liberación de gonadotropinas y la producción de esteroides ováricos.

Efectos de los esteroides gonadales cíclicos, en el tracto reproductivo
Los cambios cíclicos en la producción de estrógenos y progesterona, por los ovarios, regulan los eventos correspondientes en las trompas de Falopio, el útero, el cuello uterino y la vagina. Fisiológicamente, estos cambios preparan al útero para la implantación, y el momento adecuado de los cambios en estos tejidos, es esencial para el embarazo. Si no ocurre el embarazo, el endometrio se desprende como la descarga menstrual.
El útero está compuesto de endometrio y miometrio. El endometrio contiene un epitelio que recubre la cavidad uterina y un estroma subyacente; el miometrio es el componente del músculo liso responsable de las contracciones uterinas. Estas capas de células, las trompas de Falopio, el cuello uterino y la vagina presentan un conjunto característico de respuestas, tanto a los estrógenos como a las progestinas. Los cambios, característicos, asociados con la menstruación ocurren, principalmente, en el endometrio.
La superficie luminal del endometrio es una capa de células secretoras y ciliadas de epitelio, cilíndrico, simple, que se continúa con las aberturas de numerosas glándulas que se extienden, a través del estroma subyacente hacia el borde del miometrio. La fecundación por lo general ocurre en las trompas de Falopio, por lo que la ovulación, el transporte del óvulo fertilizado a través de la trompa de Falopio, y la preparación de la superficie endometrial deben coordinarse, temporalmente, para una implantación exitosa.
El estroma endometrial es una abundante capa de células de tejido conectivo, que contiene diversos vasos sanguíneos que experimentan cambios cíclicos asociados con la menstruación. Las células predominantes son fibroblastos, pero también están presentes macrófagos, linfocitos y otros tipos de células residentes y migratorias.
La menstruación marca el comienzo del ciclo menstrual. Durante la fase folicular (o proliferativa) del ciclo, el estrógeno comienza la reconstrucción del endometrio, estimulando la proliferación y la diferenciación. Una respuesta importante al estrógeno, en el endometrio y otros tejidos es la inducción de la PR, que permite que las células respondan a esta hormona durante la segunda mitad del ciclo.
En la fase lútea (o secretora) del ciclo, la progesterona elevada limita el efecto proliferativo de los estrógenos en el endometrio, al estimular su diferenciación. Los efectos principales incluyen la estimulación de secreciones epiteliales, importantes para la implantación del blastocisto y el crecimiento característico de los vasos sanguíneos, endometriales, observados en este momento. Estos efectos están mediados por PR-A en modelos animales.
Efectos metabólicos
Los estrógenos afectan a muchos tejidos y tienen muchas acciones metabólicas en humanos y animales. Muchos tejidos no reproductivos, incluyendo huesos, endotelio vascular, hígado, CNS, sistema inmune, tracto gastrointestinal y corazón, expresan bajos niveles de ambos ER, y la relación de ERα a ERβ varía de manera específica, de acuerdo con el tipo de célula. Los efectos de los estrógenos en determinados aspectos del metabolismo de los minerales, los lípidos, los carbohidratos y las proteínas, son de especial importancia para entender sus acciones farmacológicas. Los estrógenos tienen efectos positivos sobre la masa ósea Continuamente el hueso se remodela en sitios llamados unidades de remodelación ósea, por la acción de resorción de los osteoclastos, y la acción formadora de hueso de los osteoblastos. Los estrógenos regulan de modo directo los osteoblastos y aumentan la supervivencia de los osteocitos mediante inhibición de la apoptosis. Sin embargo, un efecto principal de los estrógenos es disminuir el número y la actividad de los osteoclastos. Gran parte de la acción de los estrógenos sobre los osteoclastos parece estar mediada por la alteración de las señales de las citocinas (tanto paracrinas como autocrinas) de los osteoblastos. Los estrógenos también aumentan la producción de osteoblastos, de la citocina osteoprotegerina (OPG), un miembro soluble, no unido a la membrana de la superfamilia del factor de necrosis tumoral. Las OPG actúan como receptores señuelo que antagonizan la unión del ligando OPG (OPG-L) a su receptor (denominado RANK, o activador del receptor de NF-κB) y evita la diferenciación de los precursores de osteoclastos a osteoclastos maduros. Los estrógenos aumentan la apoptosis de los osteoclastos, ya sea directamente o al aumentar la OPG.
Receptores de estrógenos
Los estrógenos ejercen sus efectos mediante la interacción con receptores que son miembros de la superfamilia de receptores nucleares. Los dos genes ER se encuentran en cromosomas separados: ESR1 codifica ERα y ESR2 codifica ERβ. Ambos ER son factores de transcripción nuclear dependientes de estrógenos que tienen diferentes distribuciones tisulares y efectos reguladores transcripcionales, en una gran cantidad de genes blanco.
ERα y ERβ existen como múltiples isoformas de ARNm debido al uso diferencial del promotor y a uniones alternativas. Los dos ER humanos son 44% idénticos en la secuencia general de aminoácidos y comparten la estructura de dominio común a los miembros de esta familia. Existen diferencias significativas entre las dos isoformas receptoras, en los dominios de unión al ligando y en ambos dominios de transactivación. El ERβ humano no parece contener un dominio AF-1 funcional. Los receptores parecen tener diferentes funciones biológicas y responden, de manera diferente, a varios compuestos estrogénicos. Sin embargo, su alta homología en los dominios de unión al ADN sugiere que ambos receptores reconocen secuencias de ADN similares y, por tanto, regulan muchos de los genes blanco.
El receptor de estrógenos α se expresa de forma más abundante en el tracto reproductivo femenino, especialmente, en el útero, la vagina y los ovarios, así como en la glándula mamaria, el hipotálamo, las células endoteliales y el músculo liso vascular. El ERβ se expresa por lo común, en la próstata y los ovarios, con una menor expresión en el pulmón, el cerebro, los huesos y la vasculatura. Muchas células expresan ERα y ERβ que pueden formar homodímeros o heterodímeros. Ambas formas de ER se expresan en cánceres de mama, aunque se cree que el ERα es la forma predominante responsable de la regulación del crecimiento. Cuando se coexpresa con el ERα, el ERβ puede inhibir la activación transcripcional mediada por el ERα, en muchos casos. Se han identificado variantes polimórficas de ER, pero los intentos por correlacionar polimorfismos específicos, con la frecuencia del cáncer de mama , masa ósea, cáncer de endometrio, o enfermedad cardiovascular, han dado lugar a resultados contradictorios.
Mecanismo de acción
Ambos ER son factores de transcripción activados por ligandos, que aumentan o disminuyen la transcripción de genes blanco. Después de ingresar a la célula por difusión pasiva, a través de la membrana plasmática, la hormona se une a un ER en el núcleo. En el núcleo, el ER está presente como un monómero inactivo unido a HSP90, y al unirse al estrógeno, ocurre un cambio en la conformación del ER disociándolo de las HSP que conduce a la dimerización del receptor, lo que aumenta la afinidad y la velocidad de unión del receptor al ADN. Pueden producirse homodímeros de ERα o ERβ y heterodímeros ERα/ERβ dependiendo del complemento del receptor en una célula dada. El concepto de cambios mediados por ligandos, en la conformación de ER, es fundamental para comprender el mecanismo de acción de los agonistas y antagonistas de estrógenos. El dímero del ER se une a los ERE, típicamente localizados en la región promotora de genes blanco. El complejo ER/DNA recluta una cascada de coactivadores y otras proteínas a la región promotora de los genes blanco y permite que las proteínas que componen el aparato de transcripción general se ensamblen e inicien la transcripción. Además de coactivadores y correpresores, ERα y ERβ pueden interactuar físicamente con otros factores de transcripción, como Sp1 y estas interacciones proteína-proteína constituyen un mecanismo de acción alternativo. En estas circunstancias, los complejos ER-ligando interactúan con Sp1 o AP-1 que ya está unido a su elemento regulador específico, de modo que el complejo ER no interactúa, directamente, con un ERE.
FARMACOLOGÍA
ADME (absorción, distribución, metabolismo y excreción)
Varios estrógenos están disponibles para administración oral, parenteral, transdérmica, o tópica. Dada la naturaleza lipofílica de los estrógenos, generalmente, la absorción es buena, con la preparación adecuada. Los ésteres con base acuosa o en aceite de estradiol, están disponibles para inyección intramuscular, con un rango de frecuencia que varía desde una vez a la semana, hasta una vez al mes. Los estrógenos conjugados están disponibles para la administración intravenosa o intramuscular. Los parches transdérmicos, que se cambian una o dos veces a la semana, administran estradiol de forma continua, a través de la piel. Las preparaciones están disponibles para uso tópico en la vagina o para aplicar sobre la piel. Para muchos usos terapéuticos, las preparaciones de estrógenos están disponibles en combinación con un progestágeno. Todos los estrógenos están etiquetados con declaraciones de precaución que recomiendan la prescripción de la dosis efectiva más baja y durante el tiempo más breve, compatible con los objetivos y riesgos del tratamiento para cada paciente en particular. La administración oral es común y puede usar estradiol, estrógenos conjugados, ésteres de estrona, otros estrógenos, y etinilestradiol (en combinación con un progestágeno). El estradiol está disponible en preparaciones micronizadas y no micronizadas. Las formulaciones micronizadas producen una gran superficie para una rápida absorción que supere, de forma parcial, la baja biodisponibilidad oral debida al metabolismo de primer paso.
La administración de estradiol, a través de parches transdérmicos, proporciona liberación lenta y sostenida de la hormona; distribución sistémica y niveles sanguíneos más constantes que la administración oral. El estradiol también está disponible como una emulsión tópica, que se aplica en la parte superior del muslo y la pantorrilla, o como un gel aplicado una vez al día en el brazo. La vía transdérmica no conduce a los niveles elevados del fármaco que se producen en la circulación portal después de la administración oral, por lo que se espera que minimice los efectos hepáticos de los estrógenos (p. ej., efectos sobre la síntesis hepática de proteínas, los perfiles de lipoproteínas y los niveles de triglicéridos).
MODULADORES SELECTIVOS DE RECEPTORES DE ESTRÓGENOS:
tamoxifeno, raloxifeno y toremifeno

Los moduladores de selectivos ER o SERM, son compuestos con acciones selectivas de tejido. El objetivo farmacológico de estos fármacos es producir acciones estrogénicas benéficas en ciertos tejidos (p. ej., huesos, cerebro e hígado) durante la MHT, pero con actividad antagonista en tejidos como el de mama y el endometrio, donde se producen acciones estrogénicas que podrían ser perjudiciales (p. ej., carcinogénesis). Los medicamentos de esta clase, actualmente, aprobados en Estados Unidos, so citrato de tamoxifeno, clorhidrato de raloxifeno y toremifeno, que tienen acciones similares al tamoxifeno y que están químicamente relacionados. El tamoxifeno y el toremifeno se usan para el tratamiento del cáncer de mama y el raloxifeno se usa principalmente para la prevención y el tratamiento de la osteoporosis y para reducir el riesgo de cáncer de mama invasivo en mujeres posmenopáusicas de alto riesgo.
ANTIESTRÓGENOS:
clomifeno y fulvestrant

Los compuestos antiestrógenos se distinguen de los SERM en que son antagonistas puros, en todos los tejidos estudiados. El clomifeno está aprobado para el tratamiento de la infertilidad en mujeres anovulatorias, y el fulvestrant se utiliza para el tratamiento del cáncer de mama en mujeres con progresión de la enfermedad después del tamoxifeno.
Química. Las estructuras del trans-isómero de tamoxifeno, y de raloxifeno, transclomifeno (enclomifeno) y fulvestrant son las siguientes:
El tamoxifeno es un trifeniletileno con el mismo núcleo de estilbeno que el DES, los compuestos de esta clase muestran una variedad de actividades estrogénicas y antiestrogénicas.
El raloxifeno es un compuesto no esteroideo polihidroxilado con un núcleo de benzotiofeno. El raloxifeno se une con alta afinidad tanto a ERα como a ERβ.
El citrato de clomifeno es un trifeniletileno, sus dos isómeros: zuclomifeno (cis clomifeno) y enclomifeno (trans clomifeno) son un débil agonista y un potente antagonista de estrógenos, respectivamente. El clomifeno se une tanto a ERα como a ERβ, pero los isómeros individuales no se han examinado.
El fulvestrant es una 7α-alquilamida, derivada de estradiol, que interactúa tanto con ERα como con ERβ.
EFECTOS FARMACOLÓGICOS
Tamoxifeno. El tamoxifeno exhibe actividad antiestrogénica, estrogénica o mixta, según la especie y el gen blanco medido. El fármaco tiene un efecto antirreabsortivo sobre los huesos y en los seres humanos disminuye el colesterol total, el LDL y el LPA, pero no aumenta el HDL y los triglicéridos.
Raloxifeno. El raloxifeno es un agonista de los estrógenos en los huesos, donde ejerce un efecto antirreabsortivo. Los estudios indicaron que el raloxifeno tiene un efecto antiproliferativo en los tumores de mama ER-positivos y reduce, significativamente, el riesgo de cáncer de mama ER-positivo, pero no ER-negativo.
Fulvestrant. El fulvestrant es un antiestrógeno. En ensayos clínicos, es eficaz en el tratamiento de cánceres de mama resistentes al tamoxifeno. El fulvestrant se une a ERα y ERβ con alta afinidad comparable al estradiol, pero reprime la transactivación. También aumenta drásticamente la degradación proteolítica intracelular de ERα, mientras que en apariencia protege a ERβ de la degradación.
Clomifeno. El clomifeno aumenta la secreción de gonadotropina y estimula la ovulación. Aumenta la amplitud de los pulsos LH y FSH sin cambiar la frecuencia del pulso. El efecto más destacado del clomifeno en las mujeres fue el aumento de tamaño de los ovarios y la ovulación inducida por fármacos en muchos pacientes con amenorrea, con síndrome de ovario poliquístico y con hemorragia disfuncional con ciclos anovulatorios.
USOS TERAPÉUTICOS
Cáncer de mama

El tamoxifeno es muy eficaz en el tratamiento del cáncer de mama. Se utiliza sólo para tratamiento paliativo del cáncer de mama avanzado en mujeres con tumores ER-positivos, y ahora está indicado como el tratamiento hormonal de elección para el cáncer de mama temprano y avanzado en mujeres de todas las edades.
El tamoxifeno aumenta la supervivencia libre de enfermedad y la supervivencia general. El tratamiento durante 5 años reduce la recurrencia del cáncer en 50% y la muerte en 27%, y es más eficaz
que los periodos de tratamiento más cortos de 1 a 2 años. El tamoxifeno reduce el riesgo de desarrollar cáncer de mama contralateral y está aprobado para la prevención primaria del cáncer de mama en mujeres con alto riesgo, en las que causa una disminución de 50% en el desarrollo de nuevos tumores. El tratamiento profiláctico debe limitarse a 5 años porque la efectividad disminuye a partir de entonces. El efecto secundario más frecuente son los sofocos.
Osteoporosis
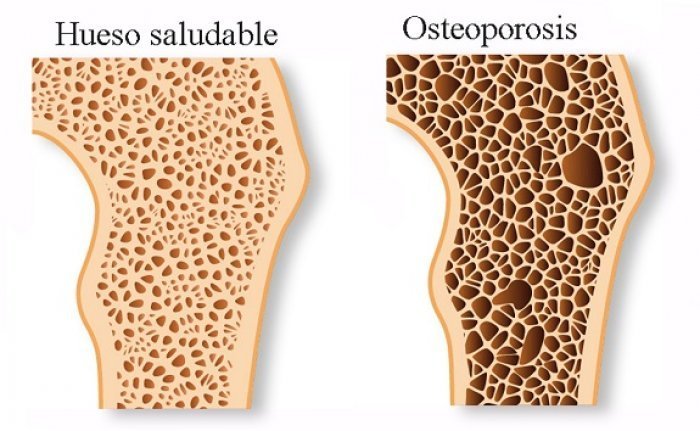
El raloxifeno reduce la tasa de pérdida ósea y puede aumentar la masa ósea en algunos sitios. En un gran ensayo clínico, el raloxifeno aumentó la densidad mineral del hueso espinal en más de 2% y redujo la tasa de fracturas vertebrales en un 30-50%, pero no redujo significativamente las fracturas no vertebrales.
Infertilidad

El citrato de clomifeno es un antiestrógeno potente que por lo general se usa para el tratamiento de la anovulación en el marco de un eje hipotalámico-hipofisiario intacto y de una producción de estrógenos adecuada (p. ej., PCOS) o para inducir la superovulación en mujeres con infertilidad inexplicada. Al inhibir los efectos de retroalimentación negativa del estrógeno, en los niveles hipotalámico e hipofisiario, el clomifeno aumenta los niveles de FSH y, por tanto, mejora la maduración folicular.
Combinaciones experimentales de SERM-estrógeno
Existe un considerable interés en MHT usando combinaciones de un agonista de estrógeno puro (p. ej., estradiol) con un SERM que tenga actividad predominantemente antagonista en la mama y el endometrio pero que no se distribuya al CNS. La estrategia es obtener las acciones beneficiosas del agonista (p. ej., prevención de sofocos y pérdida ósea), mientras que el SERM bloquee la acción agonista no deseada en sitios periféricos (p. ej., efectos proliferativos en mama y endometrio) pero sin ingresar al cerebro para causar sofocos.
PROGESTINAS
Química
Los compuestos con actividades biológicas similares a las de la progesterona se denominan progestinas, agentes progestacionales, progestágenos, progestores, gestágenos o gestógenos. Las progestinas incluyen la hormona natural progesterona, derivados de 17α-acetoxiprogesterona en la serie de pregnano; derivados de 19-nortestosterona en la serie de estrano y norgestrel y compuestos relacionados en la serie de gonano. El MPA y el acetato de megestrol son esteroides C21 en la familia pregnano, con una actividad selectiva muy similar a la de la progesterona. El MPA y la progesterona micronizada oral se usan ampliamente con estrógenos para MHT y otras situaciones en las que se desea un efecto progestacional selectivo. Además, el depósito de MPA se usa como un anticonceptivo inyectable de acción prolongada.
Biosíntesis y secreción
La progesterona es secretada por el ovario principalmente por el cuerpo lúteo, durante la segunda mitad del ciclo menstrual. La LH que actúa a través de su receptor acoplado a proteína G, estimula la secreción de progesterona durante el ciclo normal. Después de la fertilización, el trofoblasto segrega hCG en la circulación materna, que luego estimula el receptor de LH para mantener el cuerpo lúteo y la producción de progesterona. Durante el segundo o tercer mes de embarazo, la placenta en desarrollo comienza a secretar estrógeno y progesterona, en colaboración con las glándulas suprarrenales fetales y, a partir de entonces, el cuerpo lúteo no es esencial para la gestación continua. El estrógeno y la progesterona continúan siendo secretados, en grandes cantidades por la placenta, hasta el momento del parto.
ACCIONES FISIOLÓGICAS
La progesterona, producida en la fase lútea del ciclo, tiene varios efectos fisiológicos incluida la disminución de la frecuencia de pulsos de GnRH. Esta disminución, mediada por la progesterona en la frecuencia del pulso GnRH, es crítica en cuanto a suprimir la liberación de gonadotropinas y restablecer el eje hipotalámico-hipofisiario-gonadal para la transición de la fase lútea a la folicular.
Tracto reproductivo. La progesterona disminuye la proliferación endometrial, impulsada por estrógenos y conduce al desarrollo de un endometrio secretor , y la disminución abrupta de la progesterona, al final del ciclo, es el principal determinante del inicio de la menstruación. Si la duración de la fase lútea se prolonga de forma artificial, ya sea manteniendo la función lútea o mediante tratamiento con progesterona, pueden inducirse cambios deciduales en el estroma endometrial, cambios similares a los observados en el comienzo del embarazo.
Glándula mamaria. El desarrollo de la glándula mamaria requiere tanto estrógeno como progesterona. Durante el embarazo y en menor grado durante la fase lútea del ciclo, la progesterona, actuando con estrógenos, produce una proliferación de los ácinos de la glándula mamaria. Hacia el final del embarazo, los ácinos se llenan de secreciones y la vasculatura de la glándula aumenta notablemente, sin embargo, sólo después que los niveles de estrógeno y progesterona disminuyen en el parto, es que comienza la lactancia. Durante el ciclo menstrual normal, la actividad mitótica en el epitelio mamario es muy baja en la fase folicular y luego alcanza su punto máximo en la fase lútea.
CNS. Durante un ciclo menstrual normal, puede observarse un aumento en la temperatura del cuerpo basal de alrededor 0.6 °C (1 °F) a mitad del ciclo, éste se correlaciona con la ovulación. Este aumento se debe a la progesterona, pero se desconoce el mecanismo exacto de este efecto.
Efectos metabólicos. Las progestinas tienen numerosas acciones metabólicas. La propia progesterona aumenta los niveles de insulina basal y eleva la insulina después de la ingestión de carbohidratos, pero normalmente no altera la tolerancia a la glucosa. Sin embargo, la administración a largo plazo de progestinas más potentes, como el norgestrel, puede disminuir la tolerancia a la glucosa. La progesterona estimula la actividad de la lipoproteína lipasa y parece mejorar la deposición de grasa.
FARMACOLOGÍA
Mecanismo de acción
Un solo gen codifica dos isoformas de PR, PR-A y PR-B. En PR-A no se identifican los primeros 164 aminoácidos N-terminales de PR-B; esto ocurre debido al uso de dos promotores distintos en el gen PR dependientes de estrógenos. Las proporciones de las isoformas individuales varían en los tejidos reproductivos, como consecuencia del tipo de tejido, del estado de desarrollo y de los niveles de hormonas. Tanto PR-A como PR-B tienen dominios de transactivación AF-1 y AF-2, pero el PR-B es más largo y también contiene un AF-3 adicional que contribuye a su actividad celular y de promotor específica. Dado que los dominios de unión al ligando de las dos isoformas de PR son idénticos, no hay diferencia en la unión del ligando. En ausencia de ligando, el PR está presente en forma primaria en el núcleo, en un estado monomérico inactivo unido a HSP90, HSP70 y p59. Cuando los receptores se unen a la progesterona, los HSP se disocian, los receptores se fosforilan y a continuación, forman dímeros (homodímeros y heterodímeros) que se unen con alta selectividad a los PRE ubicados en genes blanco.
ADME. La progesterona experimenta un rápido metabolismo de primer paso, pero las preparaciones en dosis altas (p. ej., 100-200 mg) de progesterona micronizada están disponibles para uso oral. Aunque la biodisponibilidad absoluta de estas preparaciones es baja, pueden obtenerse niveles plasmáticos eficaces. La progesterona también está disponible en una solución inyectable de aceite como un gel vaginal; como un DIU de liberación lenta para la anticoncepción, y como un inserto vaginal para la tecnología de reproducción asistida.
ANTIPROGESTINAS Y MODULADORES DE RECEPTORES DE PROGESTERONA
Mifepristona

Química. La mifepristona es un derivado de la 19-norprogestina noretindrona que contiene un sustituyente dimetilaminofenol en la posición 11β. Compite de manera efectiva tanto con la progesterona como con los glucocorticoides por la unión a sus respectivos receptores. La mifepristona se considera un PRM debido a su actividad dependiente del contexto. Otra antiprogestina estudiada con amplitud es la onapristona (o ZK 98299), que es similar en estructura a la mifepristona, pero contiene un sustituyente metilo en la orientación 13α en lugar de 13β. Los PRM más selectivos, como el asoprisnil, se están estudiando experimentalmente.
Efectos farmacológicos
La mifepristona actúa principalmente como un antagonista del receptor competitivo para ambos PR, aunque puede tener cierta actividad agonista en ciertos contextos. Por el contrario, la onapristona parece ser un antagonista puro de la progesterona. Los complejos PR de ambos compuestos antagonizan las acciones de los complejos PR de progesterona y también parecen reclutar, de manera preferencial, correpresores. Cuando se administra en las primeras etapas del embarazo, la mifepristona causa la degradación decidual por su acción de bloqueo de los vasos sanguíneos uterinos, lo que conduce a la separación del blastocisto, disminuyendo la producción de hCG.
La mifepristona puede retrasar o prevenir la ovulación, dependiendo del momento y la forma de administración. Estos efectos se deben, en gran medida, a acciones sobre el hipotálamo y la hipófisis en lugar de los ovarios, aunque los mecanismos no están aún claros.
ADME. La mifepristona es activa por vía oral, con buena biodisponibilidad. Los niveles plasmáticos máximos ocurren al cabo de varias horas y el medicamento es eliminado poco a poco, con una t1/2 en plasma de 20-40 h. En el plasma, está unida a la glucoproteína ácida α1, que se atribuye a la prolongada t1/2 del fármaco.
Usos terapéuticos. La mifepristona, en combinación con misoprostol u otras PG, está disponible para la interrupción del embarazo en su comienzo. Cuando la mifepristona se usa para producir un aborto con medicamentos, se administra una PG 48 horas después de la antiprogestina para aumentar aún más las contracciones del miometrio y garantizar la expulsión del blastocisto desprendido.
Ulipristal

Química. El ulipristal, un derivado de la 19-norprogesterona funciona como un modulador selectivo del receptor de progesterona que actúa como un agonista parcial en PR. A diferencia de la mifepristona, el ulipristal parece ser un antagonista relativamente débil de los glucocorticoides.
Efectos farmacológicos. En dosis altas ulipristal tiene efectos antiproliferativos en el útero, sin embargo, sus acciones más relevantes hasta la fecha involucran su capacidad para inhibir la ovulación.
Usos terapéuticos. En Estados Unidos, el acetato de ulipristal tiene licencia como anticonceptivo de emergencia. Los estudios que compararon el ulipristal con el levonorgestrel (anticoncepción de emergencia sólo con progesterona) demuestran que el ulipristal es al menos igual de efectivo cuando se toma hasta 72 horas después de una relación sexual sin protección.
Beneficios de salud no anticonceptivos
Los anticonceptivos orales reducen de modo significativo la incidencia de cáncer de ovario y endometrio, al cabo de los 6 meses de uso y la incidencia disminuye 50%, después de 2 años de uso. Las inyecciones de depósito MPA también reducen sustancialmente la incidencia de cáncer uterino. Este efecto protector persiste hasta 15 años, después de suspender el uso de anticonceptivos orales. Estos agentes también disminuyen la incidencia de quistes ováricos y de enfermedad fibroquística benigna de la mama.
Contracepción poscoital
La anticoncepción poscoital (o de emergencia) está indicada en casos de falla mecánica de dispositivos de barrera o en circunstancias de relaciones sexuales sin protección. Debido a que es menos efectivo que los regímenes anticonceptivos orales estándar, no pretende ser un método anticonceptivo regular.
Interrupción del embarazo
Si la anticoncepción no se utiliza o falla, se puede usar mifepristona (RU-486) o metotrexato (50 mg/m2 por vía intramuscular u oral) para terminar un embarazo no deseado en entornos fuera de los centros quirúrgicos. A continuación, se administra una PG para estimular las contracciones uterinas y expulsar el embrión desprendido; en Estados Unidos, las PG usadas incluyen dinoprostona (PGE2) administrada por vía vaginal, o el misoprostol del análogo de PGE1, administrado por vía oral o vaginal; ambos se usan con este propósito sin que estén aprobados para ello por la FDA. Las PG usadas en otros países incluyen la sulprostona análoga de PGE2, y el gemeprost análogo de PGE1. La mifepristona (600 mg) está aprobada por la FDA para la terminación del embarazo, dentro de los 49 días posteriores al inicio del último periodo menstrual de la mujer.
INDUCCIÓN DE LA MADURACIÓN SEXUAL
Tratamiento con estrógenos en la carencia del desarrollo ovárico
En varias afecciones (p. ej., el síndrome de Turner), los ovarios no se desarrollan y la pubertad no se produce. La terapia con estrógenos, en el momento apropiado, replica los eventos de la pubertad, y los andrógenos o la hormona del crecimiento (capítulo 42), se pueden usar en concomitancia para promover el crecimiento normal. Aunque los estrógenos y los andrógenos promueven el crecimiento óseo, también aceleran la fusión epifisiaria, y su uso prematuro puede dar como resultado una altura final más corta. Los tipos de estrógenos utilizados y los regímenes de tratamiento pueden variar según el país o las preferencias individuales. Los ejemplos incluyen estrógenos conjugados, 0.3-1.25 mg; 17β-estradiol micronizado, 0.5-2.0 mg; etinilestradiol, 5-20 μg, y 17β-estradiol transdérmico, 25-50 μg. Para lograr un desarrollo mamario óptimo, el tratamiento se inicia usualmente con una dosis baja de estrógeno (p. ej., estrógenos conjugados a una dosis inicial de 0.3 mg/día, o etinilestradiol a 5 μg/día) en pacientes entre las edades de 10 y 12 años, o inmediatamente, si el diagnóstico se hace después de esta edad. Después de 3-6 meses, la dosificación se aumenta (p. ej., 0.9-1.25 mg/día de estrógenos conjugados o 20 μg/día de etinilestradiol).
Inducción de la ovulación
La infertilidad (es decir, la incapacidad de concebir después de 1 año de relaciones sexuales sin protección) afecta, aproximadamente, a 10-15% de las parejas en las naciones desarrolladas, y su incidencia es cada vez mayor a medida que más mujeres optan por retrasar el embarazo hasta periodos posteriores de su vida. La causa de la infertilidad se atribuye en principio a la mujer, en alrededor de un tercio de los casos; al hombre en aproximadamente un tercio y a ambos en cerca de un tercio. La anovulación representa aproximadamente 50% de la infertilidad femenina y es el foco principal de las intervenciones farmacológicas, utilizadas para lograr la concepción.
Clomifeno
El citrato de clomifeno fue revisado previamente en este capítulo. Un régimen típico suele ser de 50 mg/día, vía oral, durante 5 días consecutivos, comenzando entre los días 2 y 5 del ciclo, en mujeres que tienen sangrado uterino espontáneo, o después de una hemorragia inducida por la abstinencia de progesterona, en mujeres que no lo hacen. Si este régimen no induce la ovulación, la dosis de clomifeno se aumenta primero a un máximo de 100 mg/día −aprobado por la FDA−, y posiblemente a niveles más altos de 150 o 200 mg/día.
Inhibidores de aromatasa
Los inhibidores de aromatasa (p. ej., letrozol, 2.5-7.5 mg/día durante 5 días, se inician típicamente el día 3 del ciclo) inducen el desarrollo de folículos, al inhibir la biosíntesis de estrógenos, disminuyendo la retroalimentación negativa de estrógenos y aumentando los niveles de FSH y el desarrollo de folículos. Al comparar el letrozol y el clomifeno para la inducción de la ovulación en mujeres con PCOS e infertilidad, el letrozol se asoció con una mayor tasa de embarazo y nacidos vivos. El letrozol se ha asociado con menos efectos secundarios de deprivación estrógenica (sofocos, cambios de humor) y, posiblemente, menos gestaciones multifetales que el clomifeno.
Gonadotropinas
Las gonadotropinas están indicadas para la inducción de la ovulación en mujeres anovulatorias con hipogonadismo hipogonadotrópico secundario a la disfunción hipotalámica o hipofisiaria. Las gonadotropinas también se utilizan para inducir la ovulación en mujeres con PCOS que no responden a clomifeno. Dado el marcado aumento en las complicaciones maternas y fetales, asociado con la gestación multifetal, el objetivo de la inducción de la ovulación en mujeres anovulatorias es inducir la formación y la ovulación de un solo folículo dominante. Generalmente, si hay dos folículos presentes, se aceptará el mayor riesgo de gestación gemelar.
Sensibilizadores de insulina
El síndrome de ovario poliquístico afecta a 4-7% de las mujeres en edad reproductiva y es la causa más frecuente de infertilidad anovulatoria. Dado que los pacientes con PCOS a menudo presentan hiperinsulinemia y resistencia a la insulina, los sensibilizadores a la insulina, como la metformina, han sido evaluados por sus efectos sobre la ovulación y la fertilidad. Aunque varios ensayos pequeños sugirieron que la metformina aumenta la ovulación en comparación con el placebo en pacientes con PCOS, un ensayo no logró demostrar un efecto significativo de la metformina sobre la fertilidad); la metformina fue menos efectiva que el clomifeno para inducir la ovulación, promover la concepción o mejorar las tasas de nacidos vivos, y no hubo beneficio de combinar metformina con clomifeno en los nacidos vivos, excepto posiblemente en mujeres resistentes al clomifeno. Por tanto, excepto en las mujeres que presentan intolerancia a la glucosa, el consenso es que la metformina, por lo general, no debe utilizarse para la inducción de la fertilidad en mujeres con PCOS.
Este tema esta super complejo pero saber un poco de las hormonas sexuales femeninas, es muy interesante.
ResponderEliminarMe parece muy interesante la forma en la que se ha expuesto el tratamiento de los estrógenos en el desarrollo ovárico, al igual que la información en general que se plantea abiertamente, sin dudas un tema muy atrayente para conocer y estudiar,
ResponderEliminarBuen resumen. Gracias por compartir.
ResponderEliminarMuy buena informacion es bueno conocer un poco mas sobre este tema.
ResponderEliminarTema muy interesante , a veces poco conocido pero este blog ayuda mucho a entenderlo de manera oportuna.
ResponderEliminarSuper explicado e interesante. Muchas gracias por la información.
ResponderEliminarEste comentario ha sido eliminado por un administrador del blog.
ResponderEliminarGracias por la información! Muy bonito y ordenado
ResponderEliminarInteresante y muy completa información al igual está muy bien explicado.
ResponderEliminarMuy buena imformacion
ResponderEliminarInformación muy interesante y relevante.
ResponderEliminarGracias por la información.
ResponderEliminarInformación muy útil y buena.
ResponderEliminarEs de mucha utilidad esta informacion y la aplicacion de los farmacos
ResponderEliminarGracias por el dato y buen conocimiento.
ResponderEliminarConsidero que es un tema de mucha importancia al cual todas las mujeres deberían tener alcance.
ResponderEliminargracias por todo este contenido
ResponderEliminarque blog mas genial
ResponderEliminarExcelente contenido e información sobre las hormonas.
ResponderEliminarEste tema es muy fundamental para las señoritas en su ciclo muestral, ya que aquí hay una información para poder dar a saber todo sobre el sexo femenino y sus usos terapéuticos también...
ResponderEliminarExcelente manera de exponer este tema de suma importancia para la atención integral en salud de toda la población. muy buen blog!
ResponderEliminarMe gusta lo detallado de este tema, me da curiosidad saber de donde sacas la información, seria excelente ver un poco de las fuentes de donde obtienes la información pero todo lo demás está excelente.
ResponderEliminarMe gusta el tema y esta muy bien detallado todos los puntos
ResponderEliminarExcelente información
ResponderEliminarExcelentes aportes, y muy buena distribución de la información!!!
ResponderEliminarUn tema muy interesante, muy comprensible y organizado, excelente trabajo.
ResponderEliminar